The AI Prescription: Separating Promise from Performance in Drug Development
By Dr Tamar Raz Life Science Innovation & Commercialization Executive
The year 2024 marked a historic turning point in the recognition of AI, as the Nobel Committee awarded not one, but two prestigious prizes to AI pioneers whose work is revolutionizing medicine and scientific discovery.
The Nobel Prize in Physics was awarded to John J. Hopfield of Princeton University and Geoffrey E. Hinton of the University of Toronto for their groundbreaking development of machine learning technology using artificial neural networks. Hinton, often revered as the “AI Godfather,” laid the foundational stones for the neural networks that power today’s AI revolution.
Simultaneously, the Nobel Prize in Chemistry recognized the transformative power of AI in solving one of biology’s most enduring mysteries. Demis Hassabis and John M. Jumper of Google DeepMind, alongside David Baker of the University of Washington, were honored for their breakthrough in protein structure prediction and design. Their creation of AlphaFold2 solved a 50-year-old scientific puzzle, enabling researchers to predict the three-dimensional structures of proteins with unprecedented accuracy.
These Nobel recognitions signal more than academic achievement – they indicate the current era where AI becomes an indispensable tool in medical research, drug discovery, development of new treatments, our understanding of diseases, and fundamentally transform how we approach healthcare challenges in the 21st century.
By July 2025, the FDA’s AI device registry had grown to approximately 1,247 approved clinical AI algorithms, with roughly 300 new clearances granted in the preceding year alone, a pace of adoption that underscores both the accelerating integration of AI into medical practice and the urgency of addressing the regulatory and safety challenges that accompany such rapid deployment.
However, while holding a transformative promise for healthcare, AI integration into medical practice faces significant obstacles and potential dangers that must be carefully addressed, the main concern include:
Data and Algorithmic Challenges
AI systems trained on biased datasets can preserve and amplify existing healthcare disparities. These biases can lead to substandard clinical decisions and worsen longstanding inequalities in healthcare outcomes among different demographic groups. This problem is compounded by AI trained on unstructured or biased data that might generate misleading results.
Patient Safety and Medical Errors
The most immediate concern involves direct patient harm. AI system errors pose risks of injuries to patients. Healthcare providers face the additional challenge of automation bias, where humans become overly reliant on AI systems despite their cognitive limitations, potentially leading to critical oversights in patient care.
Privacy and Data Protection
The digital transformation brings unprecedented risks to patient confidentiality. AI generates vast amounts of sensitive patient data, creating data privacy and security risks. More concerning, AI also magnifies existing cyber-security risks, potentially threatening patient privacy and confidentiality.
Implementation and Adoption Barriers
The path from laboratory to clinic presents multiple obstacles. Translating AI systems into healthcare include issues inherent to machine learning, logistical implementation hurdles, and adoption barriers. Healthcare providers struggle with concerns about data quality, bias, privacy, and accuracy of models, while many institutions lack the technical expertise needed for proper implementation.
The Regulatory Challenge
While over 1,000 AI/ML-enabled medical devices and technologies have been authorized by the FDA as of early 2025, regulatory agencies struggle to keep pace with technological advancement. The rapid deployment often outpaces comprehensive safety testing and standardization efforts.
In summary – while AI promises to revolutionize healthcare, its implementation faces significant obstacles that could undermine patient safety and healthcare equity. Success requires addressing these challenges proactively through improved data governance, bias mitigation strategies, enhanced cybersecurity measures, and comprehensive regulatory frameworks that balance innovation with patient safety.
The Israeli Perspective
Israel has emerged as a global leader in AI-powered healthcare innovation, with companies developing cutting-edge solutions that directly address many of the hurdles and risks discussed above. Here are five examples of how Israeli AI medical companies are tackling these challenges:
Aidoc – AI-Powered Medical Imaging for Critical Care: Aidoc is helping medical centers alert imaging technicians with AI-based insights of possible bleeding in the brain and other critical conditions in a patient’s scan within minutes. This directly addresses the challenge of integrating AI into clinical workflows without disrupting existing practices.
Ibex – AI-Powered Diagnostic Solutions: Ibex has led the way in AI-powered diagnostics for pathology, helping pathologists ensure better cancer care for patients around the world. Developed by pathologists for pathologists, Ibex solutions serve the world’s leading physicians, healthcare organizations and diagnostic providers.
MDClone – Revolutionary Privacy-Preserving Analytics: MDClone offers a new healthcare data paradigm, enabling fast and direct access to healthcare data while fully protecting patient’s privacy. The company addresses one of the most critical challenges in medical AI: accessing valuable healthcare data without compromising patient confidentiality.
CytoReason – AI-Driven Drug Development: CytoReason, an Israeli startup that uses artificial intelligence to develop computational disease models for drug discovery, secured funding backed by US chipmaker Nvidia and pharma giant Pfizer. This addresses the challenge of developing more effective and representative medical treatments.
Rhino Health – Privacy-Preserving Collaborative AI: Rhino Health is using federated learning to make AI development more collaborative while maintaining privacy. This innovative approach addresses both data sharing limitations and privacy concerns.
These examples represent only a small portion of Israel’s robust medical AI sector. Collectively, they and many other Israeli companies illustrate that the significant theoretical challenges facing medical AI can be effectively addressed through innovative design approaches and close alignment with actual clinical needs and workflows.
About the author
Dr Tamar Raz
Life Science Innovation & Commercialization Executive
Tamar Raz, PhD, is a life sciences executive with 20+ years of experience in business development, alliance management, and technology transfer. Formerly CEO of Hadasit, she built global partnerships, executed high-value licensing deals, and co-founded the Hadassah Accelerator with IBM. She currently serves as Chief Strategy Officer at EMRIS Pharma, advising on growth and strategic collaborations. Profile: https:linkedin.com/in/tamar-raz/
Opportunities and Regulatory Challenges for Healthspan Therapeutics
By Thomas Seoh CEO of Kinexum and EVP of the Kitalys Institute
The longevity biotechnology sector is experiencing increasing momentum. Therapeutic interventions targeting healthspan – the period of life spent in good health – are transitioning from basic research to clinical translation, and may well start to be commercialized this decade.
The Minovia Therapeutics SPAC deal, announced June 25, 2025, which values the Haifa-based company at $180 million pre-money, targets rare mitochondrial diseases and age-related decline. Its leadership is connected with Israel’s premier research institutions, including the Hebrew University, the Weizmann Institute, the Israel Institute for Biological Research, and Sheba Medical Center.
The $101 million+ XPRIZE Healthspan challenges competitors to develop therapeutic treatments that significantly extend healthy lifespan by 10 or 20 years by restoring muscle, cognitive, and immune function in individuals aged 65-80, within a treatment period of one year. This unprecedented prize pool has been assembled to incentivize the clinical development of highly robust aging interventions on actionable timelines.
ARPA-H’s PROSPR program in the US seeks contract parties to develop an Intrinsic Capacity composite score measurable at home that predicts long-term health outcomes based on physiological and biochemical measures, and repurposed or new drugs that increase healthspan. Unlike traditional disease-focused initiatives, PROSPR explicitly targets the pre-disease state, where geroscience research suggests the maximum impact for increasing healthspan.
This emerging sector, projected to become a trillion-dollar industry, faces a critical challenge: regulatory frameworks designed for traditional disease-specific therapeutics are not well-equipped to evaluate products targeting mechanisms in the biology of aging.
The Geroscience Revolution
Geroscience – the interdisciplinary field examining the intersection between aging biology and chronic diseases and health – has revealed that aging is not merely an inevitable decline but a malleable biological process. Evidence has been accumulating at an accelerating pace that targeting fundamental aging mechanisms may be able to simultaneously influence multiple age-related conditions, including cardiovascular disease, cancer, neurodegeneration, and metabolic disorders.
This scientific paradigm shift challenges the traditional “one drug, one disease” model that has dominated allopathic pharmaceutical development for decades. Instead of treating individual chronic diseases after they manifest, healthspan therapeutics aim to address their common underlying driver: aging itself.
Regulatory Roadblocks
The regulatory challenges facing healthspan therapeutics are structural and cultural. If a company today approaches the FDA claiming to have developed a pill that “cures aging,” this is unevaluable under current regulations.
Aging is not classified as a disease by regulatory agencies. FDA typically approves treatments for specific diseases in defined patient populations. Healthspan products target multiple chronic diseases across heterogeneous populations.
Moreover, the FDA typically requires a demonstration of clinical benefit in how patients ‘feel, function, or survive.’ This a fortiori limits evaluation of interventions to symptomatic disease states, and creates barriers for upstream interventions targeting molecular or cellular changes in pre-disease states – precisely where geroscience research suggests the greatest impact lies.
Further, vaccines, statins and contraceptives are exceptions that prove the rule, that FDA is not culturally well-equipped to evaluate a preventive therapeutic intervention for healthier, younger people who are not yet patients, and don’t want to be.
And, the timeframes required to demonstrate improved healthspan or longevity in human studies can span decades, creating practical and financial barriers that the private sector typically does not have the ability or stomach to overcome.
Clinical Development Complexities
Beyond regulatory hurdles, healthspan therapeutics face unique clinical development challenges. Traditional clinical trial designs, optimized for acute treatments in symptomatic patients, are poorly suited for interventions targeting healthy aging.
Biomarker validation represents a critical bottleneck. While researchers have identified numerous molecular signatures of aging, translating these into acceptable surrogate endpoints for regulatory development remains a multi-year or multi-decade challenge. The field lacks standardized, validated biomarkers that regulatory agencies will accept as meaningful indicators of healthspan improvement.
Patient recruitment poses additional complexity. Unlike traditional disease-focused trials that enroll patients with clear symptoms, healthspan studies must identify and enroll healthy individuals at risk for age-related decline – a population that may lack motivation or urgency to participate in lengthy clinical trials.
The heterogeneity of aging also presents statistical challenges. Individual variations in genetic background, lifestyle factors, and environmental exposures create substantial noise in clinical outcomes, requiring larger study populations and longer observation periods than traditional trials.
Looking Ahead
The THRIVE Act,* developed by the Kitalys Institute (www.kitalys.org), exemplifies a proposed solution to the regulatory challenge. This draft legislation would establish an optional regulatory pathway, escalating evidentiary requirements and incentives for therapeutic interventions, devices, and supplements targeting healthy longevity. A THRIVE Act version 2.0, incorporating feedback from multiple stakeholders, is now available at www.kitalys.org.
The convergence of advancing geroscience, increasing commercial interest, boosted by GLP-1 drugs, and healthy longevity entering mainstream conversations is driving healthspan products from experimental to mainstream medicine. As the field matures, we can expect standardized biomarkers, novel clinical trial designs, evolving regulatory, reimbursement, and other policies, a multitude of different approaches to increasing healthspan,** and the continuing explosion in longevity medicine. Regulation and policy can accelerate or delay the transition from “sick-care” to precision health, but forward-thinking companies will look for opportunities to get ‘ahead of the curve’ in this inevitable paradigm shift.
* The draft THRIVE Act proposes an optional regulatory pathway for healthspan products (those that can prevent, delay, reduce the risk of, or reverse chronic diseases of aging), with three tiers of escalating evidentiary requirements, and a period of market exclusivity per tier, to incentivize the generation of clinical evidence for extending healthspan. Tier 3 is analogous to full FDA approval, Tier 2 somewhat analogous to Accelerated Approval, and Tier 1 would afford qualifying products earlier access to the market than under current regulations, in order to generate increasingly rigorous clinical data on the capitalization of a commercial-stage company.
About the author
Thomas Seoh
CEO of Kinexum and EVP of the Kitalys Institute
Kinexum is a regulatory, clinical, product and corporate development strategic advisory firm (www.kinexum.com). Since 2017, Kinexum, and later its not-for-profit Kitalys Institute (www.kitalys.org), have organized the Targeting Healthy Longevity (previously called Targeting Metabesity) conference, convening leaders of NIH, FDA, Congress, the UK Parliament, geroscience and chronic disease research, industry and capital markets in furtherance of the Kitalys mission to accelerate the translation of science into public health to prevent chronic diseases and extend healthy longevity for all. Over 200 conference sessions are posted on Kitalys’s YouTube channel at www.healthy-longevity.org. Kitalys has advised XPRIZE Healthspan and ARPA-H PROSPR on strategic regulatory matters, and Kinexum represents a number of longevity biotech companies, including semi-finalists in XPRIZE Healthspan.
Clinically validated digital therapeutics in oncology
By Daniel Israel President
In recent years, digital health technologies have been increasingly integrated into oncology, from patient-reported outcomes (PROs) to AI-driven decision support. One of the main challenges is ensuring that these solutions are supported by robust, long-term clinical evidence. Demonstrating not only usability but also measurable effects on survival and quality of life is essential before these tools can be considered part of standard cancer care.
Among the digital therapeutics evaluated to date, we can take the example of Moovcare®, which has accumulated more than a decade of clinical research across randomized trials and real-world studies in France, Spain, and the United States. Its approach is based on a simple principle: patients complete a weekly questionnaire, developed by physicians. Data are analyzed by an algorithm designed to detect early signs of relapses or complications. Notifications are then transmitted to a medical team, who reviews the information and, when necessary, contacts the patient. If a medical intervention is required, the oncologist is informed so that appropriate action can be taken before the patient’s condition deteriorates. It has already been used by thousands of patients. This approach complements conventional imaging and routine visits, supporting more timely clinical management.
Studies have shown an increase in overall survival of 7.6 months compared with standard follow-up, alongside improved quality of life, fewer hospitalizations, and adherence rates above 90%. Economic analyses also point to reductions of more than 10% in annual hospitalization costs. These results have been presented at international oncology congresses such as ASCO and published in leading peer-reviewed journals including JNCI, JTO, and JAMA.
In 2025, Moovcare® obtained CE Mark certification under the European Regulation (EU) 2017/745 for Moovcare® Toxicity, designed to monitor treatment-related toxicities across all cancer types. At the same time, Moovcare® Lung renewed its CE certification. This dual recognition reflects both regulatory compliance and the maturity of clinical validation achieved by the solutions.
Clinical practice feedback aligns with the trial results. Physicians report that this tool offers a structured approach to capturing patient-reported outcomes and supports earlier therapeutic decision-making. Patients describe the tool as easy to use and reassuring, with consistently high satisfaction across studies. Integration into clinical workflows has been feasible without compromising safety or oversight.
The broader lesson is clear: digital health in oncology can deliver real value. Long-term evaluation, regulatory approval, and integration into care pathways are critical for adoption at scale.
Moovcare® illustrates this pathway from concept to validated digital therapeutic. It is now entering a phase of international expansion. Its trajectory highlights how digital monitoring, grounded in robust evidence, can contribute to earlier relapse detection, better quality of life, and more efficient use of healthcare resources in cancer care.
About the author
Daniel Israel
President
Short bio : Daniel Israel is the President and founder of Sivan Innovation, where he leads the development of Moovcare®, the first clinically validated digital therapeutic for cancer. With nearly 20 years in digital health, he previously founded Sephira Group, whose flagship is the leading platform in France for secure billing and data transmission between patients, providers, and payers. https://www.moovcare.com/
Why, What, How to Interpret Machine Learning Models in Medical Research
By Wooyoung Kim, Ph.D Department of Computing and Software Systems School of STEM University of Washington Bothell
What are your feelings on AI (Artificial Intelligence) and ML (Machine Learning)? Are you excited, like waiting for a new video game or the latest iPhone release? Or are you fearful or concerned? If it’s the latter, what exactly worries you? Specifically, if you work in biology, bioinformatics, health informatics, or medical research, what aspects of AI/ML are most concerning? Perhaps it is because AI/ML applications in these fields feel more impactful than in others, as the decision can be life-changing. You might worry about losing your job, fearing that you can not compete with machines. Or you may be concerned about not being able to detect incorrect results produced by automated systems. You could also be experiencing the “Uncanny Valley” phenomenon in AI: people generally like human-like systems at first, but once they become almost human, the comfort levels drop sharply, leaving people uneasy or even repulsed.
However, we cannot avoid AI/ML simply because we are afraid or feel uncomfortable with it. Instead, we should dive deeper into this “magical” field, learn to understand it well enough to overcome our fears, and challenge any incorrect assumptions. The most dangerous mistake is to trust a machine without truly understanding how it works. So, let’s begin by exploring how these systems function, especially in the context of medical research.
Artificial Intelligence (AI) is a broad concept referring to any computer system designed to simulate human intelligence. In medical research, AI can include clinical decision support tools, automated diagnostic systems, or technologies that read and process medical records. Machine Learning (ML) is a subset of AI that focuses on learning patterns from data without relying on hand-crafted rules or explicit programming. ML is used in medical research to predict or diagnose disease risk from patient data, detect diseases from medical images, and identify biomarkers from genomic data.
Roles of ML in Medical Research
In this article, I will focus on machine learning (ML) in medical research, as artificial intelligence (AI) is broader and is outside my area of expertise. So, what is ML in general? ML is the process of discovering patterns in data to derive rules that can be applied to new datasets.
Imagine you want to predict whether a patient has cancer based on their symptoms, demographic information, and responses to a questionnaire, but without access to biopsy results for confirmation. The typical clinical process is to identify potential signs based on prior experience or medical knowledge, then proceed with further testing such as blood work, ultrasound, and eventually, biopsy. As you can imagine, this process requires a significant amount of time and relies heavily on the doctor’s careful examination and decision-making.
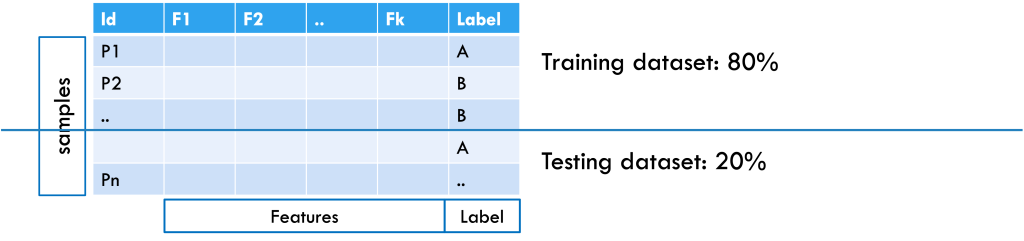
Here’s the process and role of ML in this decision-making context: First, data are collected from people who have already been diagnosed with cancer by doctors, as well as from people who are healthy or have other conditions. Each person is considered a sample, and each piece of information collected, such as age, blood test results, or scan findings, is called a feature. The dataset also includes whether each person has cancer or not, which serves as the label or output, as shown in Figure 1.
Figure 1: Example of a patient dataset formatted for machine learning, with an 80% training and 20% testing split. Samples are arranged in rows, features in columns, and the class label is included.
After collecting the data, it is split into training and testing sets (for example, 80% and 20%). The training set helps the machine learning system learn patterns in the features that separate cancer patients from non-cancer patients. If the dataset is large enough, the training set can be further split into a training subset (for learning the model) and a validation subset (for tuning hyperparameters and selecting the best model). To check how well the model works, it is tested on the testing set, where the labels are hidden. The model makes predictions based on what it learned, and these predictions are compared with the actual labels to measure accuracy. This approach considers all features together, since a single piece of information is usually not enough for a diagnosis. From the data, the model learns rules; for example, “A combination of elevated WBC, low platelets, and anemia suggests a 90% chance of cancer.” Once tested, the model can be applied to new patient samples to provide predictions.
Challenges in ML in medical research
What can go wrong? Well, every step can go wrong, and each step involves its own set of challenges. Applying ML to any research generally requires several key steps: defining a clear goal; collecting and preprocessing a dataset aligned with the goal; applying ML algorithms and evaluating them to select proper ones; and translating the results into a practical application. To illustrate these challenges, consider a case study to diagnose Merkel Cell Carcinoma (MCC), a rare and aggressive skin cancer [1]. Because MCC is difficult to diagnose early, even for experts, and its prognosis is equally challenging, researchers may turn to ML techniques to aid the diagnostic or prognostic process. This immediately raises a fundamental question: What kind of ML task is this?
Which ML task? Translating clinical questions into machine learning (ML) problems is not always straightforward. If your goal is early diagnosis of MCC, then it is a classification task, as you aim to classify cases as either MCC (positive class) or not (negative class). On the other hand, if your goal is to predict how much the tumor will grow over time to guide treatment decisions, that is, to prognose MCC, then it may be framed as a regression problem.
Additional complexity depends on the nature and availability of your dataset. If your data includes labeled examples (e.g., cancer vs. non-cancer for classification, or tumor size measurements over time for regression), then you are dealing with a supervised learning problem. However, if your dataset lacks labels, then it may fall under unsupervised learning.
ML-ready dataset Assuming a supervised classification task for early MCC diagnosis, the next challenge is assembling an ML-ready dataset. Ideally, this would include more than 1,000 complete and clean samples with balanced classes (MCC vs. non-MCC). In practice, MCC data rarely meet these conditions.
A key hurdle is defining the negative or control class. If controls are only healthy individuals, the model may simply learn “disease vs. no disease” instead of MCC-specific patterns. If controls are patients with non-cancer conditions, the task shifts to “cancer vs. non-cancer.” Including other skin cancers as controls creates a more targeted but difficult comparison. The choice of controls must therefore align with the project’s goal.
Data scarcity and inconsistency further complicate the problem. Since MCC is rare, data can be fragmented across institutions, and ethical or legal restrictions would limit access. Therefore, available datasets are typically small, imbalanced, and noisy, with inconsistent features due to varying diagnostic practices. Imbalance is particularly harmful: For instance, in a dataset of 500 samples (50 positives, 450 negatives), a split leaves only 10 positives in the test set. A model predicting all cases as negative would achieve 90% accuracy yet fail to detect any positives. This is the case where accuracy becomes misleading, and the dataset is unreliable for ML.
What is the ‘Right’ model? Some models are easy to understand, as they provide the rules in a human way. For example, Regression Models provide the model as a linear equation that adds each feature with their corresponding weight, and with a Decision Tree model, you can derive the rules that split to the next branch based on conditions and rules, and finally reach your decision. In those cases, the model itself can be interpretable, and you can evaluate the model with their domain contexts. Those interpretable ML models are referred to as White-box models [2]. However, they generally work well with certain assumptions, such as linear interactions of features, independence of features, or a small number of features. Therefore, in the process of data preprocessing, you could lose critical information or interfere with the interpretation of the predictions.
As ML develops, more computationally solid and complex models have been developed, including ensemble models, and it generally provides better results with standard evaluation metrics – e.g., accuracy, precision, or sensitivity. Since healthy and medical data are often longitudinal or irregular over time, in this case, you might need to use time-aware models, for example, RNNs, transformers, or survival models. However, those models are generally referred to as Black-box models, which hide the intrinsic structures and obstruct translating the results to their domain applications, leading to skepticism in the models.
Translating the results Now, with the trained model, you want to predict if a new patient will be correctly diagnosed with the disease by providing the patient’s information to the model. However, it just provides the predicted outcome with certain (e.g., 90%) accuracy. What if the outcome does not match your domain knowledge, or the outcome seems incorrect? You will ask “why?” with some following questions: Were the predictions made without bias?; are they reliable and safe? How were the predictions, and how does the model work?
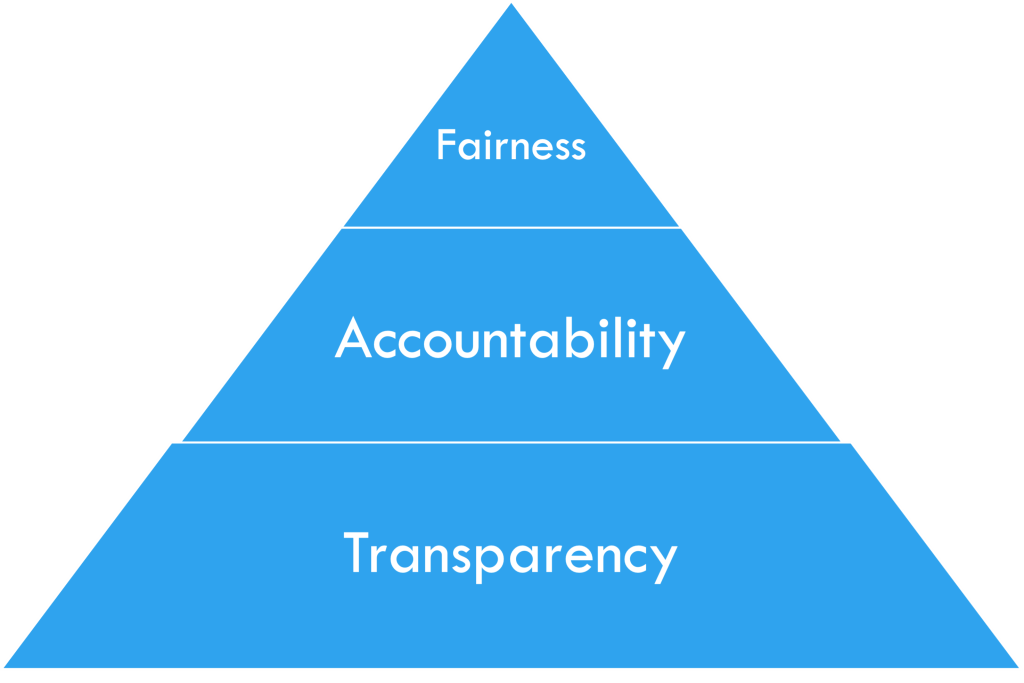
These questions lead to the main concepts of interpretable machine learning (iML), FAT: Fairness, Accountability, and Transparency.
Figure 2: FAT in iML, by courtesy of [2]
iML in medical research
Interpretable Machine Learning (iML) is a developing field that seeks to address the questions of Fairness, Accountability, and Transparency (FAT) [2]. The FAT concepts are often organized in a pyramid form, as in Figure 2, with Fairness at the top and Transparency at the bottom, as each layer builds upon the previous one. In this section, I will discuss the ethical considerations of applying ML techniques to medical research and examine how they relate to each concept of FAT [3].
Fairness in medical research As a starting point in iML, the ethical consideration of fairness comes from the principle of Justice: Basically, it is asking whether the predictions are made without bias, and when applying ML in medical research, the following are the key fairness issues [4]:
Representation bias: Is the dataset fair? Are certain subgroups (e.g., age, race, gender, or rare diseases) underrepresented?
Labeling bias: Does the dataset include delayed or missed diagnoses? Are there misclassifications?
Measurement or instrument bias: Do different labs use slightly different testing setups that could introduce variation?
Evaluation bias: Are error rates equal across groups? Are evaluation metrics fairly applied and able to capture all necessary concerns?
Consider the MCC case as an example. It may show clear representation bias, since MCC cases are underrepresented. Some test results may be missing due to differences in insurance policies or patients’ financial constraints. Different practitioners may order different lab tests, further affecting representation. Labeling bias can occur when patients have different outcomes across timelines, as rare diseases often take time to be detected, and test results unrelated to MCC may look similar, which could add ambiguity. Measurement bias may arise from data collected across multiple hospitals with varying protocols. Finally, evaluation bias often follows from representation bias: if accuracy is used as the main metric, it may fail to capture the true detection rate for underrepresented classes.
Accountability in medical research Once fairness is addressed, accountability is grounded in the ethical principle of Responsibility: Who is responsible for errors or harm caused by ML predictions or the decisions informed by them? In iML, accountability highlights several key issues in medical research as follows [5, 6, 7]:
Responsibilityandliability: Who is accountable if harm occurs?
Model reliability and safety: Does the model consistently provide the same outputs across populations or subgroups? Is its performance stable over time?
Data quality and label integrity: Can we trust the model’s predictions if labels are temporally inconsistent? Is there data or target leakage, where a proxy for expert decisions unfairly influences model outcomes?
Consider the MCC case in terms of accountability. If the model misclassifies rare MCC cases, who should be blamed? Clinicians who used and trusted the system? ML developers who built it? Or data curators who prepared the dataset? Reliability may also be questioned if the model performs inconsistently across hospitals, especially when testing protocols differ. Another challenge is label integrity: MCC diagnoses can change over time, and models may suffer from target leakage, where certain features are influenced by expert curation rather than independent data.
Transparency in medical research Alongside fairness and accountability, transparency is tied to the ethical principle of Trust: Without sufficient transparency, trust in ML systems used in medicine can be compromised, and the key issues in transparency include the following [8, 9, 10, 11, 12, 13]:
Model interpretability: Are the inner workings of the model understandable to clinicians, patients, and regulators? Can predictions be explained in a clinically meaningful way?
Decision traceability: Can each prediction be traced back to the data and reasoning process that produced it? Is there an auditable record of model outputs?
Communication of limitations: Are the model’s constraints, risks, and intended uses clearly communicated to all stakeholders? Are uncertainty estimates provided with predictions?
Accessibility of information: Is sufficient documentation, such as data sources, pre-processing steps, and model validation results, made available for evaluation?
Consider the MCC case in terms of transparency. Since MCC is rare and often underrepresented, even a high-performing model may be opaque and difficult to interpret. Preprocessing steps can further obscure decision pathways, making it hard to trace why a prediction was made. Without clear communication between developers and clinicians about the model’s limitations and risk factors, it cannot be reliably adopted for life-critical decisions. Ultimately, transparency is essential to ensure safe and trustworthy access to information.
iML: Why, What, and How?
Why iML in medical research? Because machine learning (ML) alone is not sufficient in medical research, where decisions can be life-changing, iML is essential to ensure that models are both understandable and trustworthy. It is important to recognize that current iML techniques are limited in many ways and are mostly developed for general applications. Their direct use in medical and health research requires caution and, in some cases, the development of new domain-specific methods. Rather than focusing only on applying existing techniques, it is crucial to emphasize the underlying ethical principles of fairness, accountability, and transparency (FAT).
What is iML in medical research? Ideally, its focus is on interpretation without sacrificing performance. This allows it to reduce the risk of incorrect decisions, trace the reasoning behind decisions, and provide explanations for predictions in medical research. However, iML is still in its early stages, with few established standards, limited case studies, and only a handful of success stories in medicine. These factors make research on interpretable ML especially challenging, since interpretability must be demonstrated rather than assumed. Therefore, iML in medical research should include practical guidelines for use and proper evaluation metrics that measure not only performance but also
interpretability within the given context. Because decisions can be life-changing, iML in medical research is closely tied to ethics and regulations, requiring a broader and more practical understanding of interpretation.
Then how can iML be applied in medical research? First, current iML techniques can be leveraged, most of which help identify features that drive predictions. They can also generate counterfactuals that show what might change outcomes and provide human-readable rules to explain results. In our MCC case study, for example, the top-ranked feature might be a blood test or MRI result, which could serve as a valuable clinical guideline. At the individual patient level, counterfactuals can reveal what factors might have led to a different prediction, offering practical insights for prevention and early intervention. These functions embody fairness by clarifying which variables influence decisions, strengthen accountability by making model behavior examinable, and promote transparency by offering explanations in human-understandable terms.
Researchers must also be explicit about limitations and risks, supported by detailed documentation to ensure reproducibility and oversight. Since AI relies on vast and constantly evolving information sources, the reliability and timeliness of those sources are critical. Responsibility for trustworthy data extends beyond AI developers to the broader research community. Thus, monitoring, investigating, and developing better ways to maintain reliable data, while preventing the spread of misinformation, are also the researcher’s responsibility. Because outcomes directly affect human lives, especially in medical research, regulations guided by FAT principles should govern data collection, model use, and result interpretation from the outset. Without such safeguards, technological advances risk outpacing both ethical governance and clinical responsibility.
So, are you ready to bring AI and ML into your research?
References
J. C. Becker, A. Stang, J. A. DeCaprio, L. Cerroni, C. Lebb´e, M. Veness, and
P. Nghiem, “Merkel cell carcinoma,” Nature Reviews Disease Primers, vol. 3, p. 17077, 2017.
S. Mas´ıs, Interpretable machine learning with python. Packt Publishing Birming-ham, UK, 2021.
T. L. Beauchamp and J. F. Childress, Principles of biomedical ethics. Edicoes Loyola, 1994.
N. Mehrabi, F. Morstatter, N. Saxena, K. Lerman, and A. Galstyan, “A survey on bias and fairness in machine learning,” ACM computing surveys (CSUR), vol. 54, no. 6, pp. 1–35, 2021.
D. S. Char, N. H. Shah, and D. Magnus, “Implementing machine learning in health care—addressing ethical challenges,” The New England journal of medicine, vol. 378, no. 11, p. 981, 2018.
T. Gebru, J. Morgenstern, B. Vecchione, J. W. Vaughan, H. Wallach, H. D. Iii, and K. Crawford, “Datasheets for datasets,” Communications of the ACM, vol. 64, no. 12, pp. 86–92, 2021.
H. Suresh and J. V. Guttag, “A framework for understanding unintended conse-quences of machine learning,” arXiv preprint arXiv:1901.10002, vol. 2, no. 8, p. 73, 2019.
S. G. Finlayson, A. Subbaswamy, K. Singh, J. Bowers, A. Kupke, J. Zittrain, I. S. Kohane, and S. Saria, “The clinician and dataset shift in artificial intelligence,” New England Journal of Medicine, vol. 385, no. 3, pp. 283–286, 2021.
J. A. Kroll, “Accountable algorithms,” Ph.D. dissertation, Princeton University, 2015.
F. Doshi-Velez and B. Kim, “Towards a rigorous science of interpretable machine learning,” arXiv preprint arXiv:1702.08608, 2017.
C. Rudin, “Stop explaining black box machine learning models for high stakes de-cisions and use interpretable models instead,” Nature machine intelligence, vol. 1, no. 5, pp. 206–215, 2019.
E. Begoli, T. Bhattacharya, and D. Kusnezov, “The need for uncertainty quantifi-cation in machine-assisted medical decision making,” Nature Machine Intelligence, vol. 1, no. 1, pp. 20–23, 2019.
C. J. Kelly, A. Karthikesalingam, M. Suleyman, G. Corrado, and D. King, “Key challenges for delivering clinical impact with artificial intelligence,” BMC medicine, vol. 17, no. 1, p. 195, 2019.
About the author
Wooyoung Kim, Ph.D
Department of Computing and Software Systems School of STEM University of Washington Bothell
Dr. Wooyoung Kim is an Associate Professor in the department of Computing and Software Systems at the School of STEM, University of Washington Bothell. She earned her Ph.D. and M.S. degrees in Computer Science from Georgia State University, and Georgia Institute of Technology, Atlanta, GA., in 2012, and 2007 respectively. Her research interests lie in bioinformatics and systems biology, an interdisciplinary study field that integrates various areas of computer science - including machine learning, data mining, numerical analysis, graph theory and pattern recognition - with subjects such as biology, chemistry, engineering, statistics, health-informatics, and medical research.
Using AI for Regulatory Referencing in CMC Consulting: Risks and Mitigations
By Tamar Oved QA and CMC Director at ADRES Advanced Regulatory Services
As a consultant supporting CMC development during clinical stages, I regularly use AI tools to enhance a range of tasks – from drafting CMC development plans and setting specifications, to understanding analytical testing requirements and identifying relevant regulatory guidelines. When applied thoughtfully, AI can significantly streamline complex and time-consuming processes. However, its use also introduces specific risks that need to be carefully managed to ensure reliability, compliance, and scientific integrity.
One major concern is the use of outdated or inaccurate content. As is probably already known, AI models are typically trained on historical data and may not reflect the most current versions of regulatory guidelines. For example, if I ask the AI about ICH Q2, it might reference an earlier draft or fail to incorporate recent Q&A updates. For example, when I used ChatGPT for a specific question relating to ICH Q2 (Validation of analytical procedures), even when I used its agent “WEB pilot”, which should access the most recent versions on the Internet, the answer I got was incorrect since it was based on version 1 instead of version 2 of the guideline. The risk is even higher for region-specific guidance – AI may inadvertently cite FDA expectations for an EMA dossier or vice versa. To mitigate this, I always verify outputs directly against official sources like EMA and FDA guidelines.
Another issue is source transparency. AI models can provide generalized summaries without clear citations or links to source documents. This lack of traceability is problematic in a regulated context, especially when we need to justify decisions during inspections or in Module 3 submissions. I’ve learned to treat AI outputs as pointers, not definitive sources, and as mentioned above, I always follow up by locating the original document myself.
There’s also the risk of hallucinated references-fabricated documents or clauses that sound convincing. These are dangerous in any regulatory context. I’ve seen AI create entirely fictional guidance sections or merge unrelated guidelines into one. As above, cross-referencing with validated regulatory databases (like RAPS, Cortellis, or agency portals) or relevant guidelines (EMA, FDA, ICH) helps ensure credibility. Additionally, I have noticed that some AI tools are less ‘hallucinating’ than others. E.g., Perplexity AI is usually more accurate. Alternatively, specific agents in ChatGPT (e.g., ‘ChatGMP’ and ‘Medical Device Regulatory Advisor’) are also usually more accurate.
A less obvious but serious risk is, of course, data protection. If I’m consulting on proprietary projects and use cloud-based AI tools that aren’t validated or private, there’s potential exposure of confidential sponsor data. This could breach both GDPR and confidentiality agreements. My solution: I never put sensitive information into unsecured platforms and additionally use enterprise-grade tools with appropriate data controls.
Last but not least, AI lacks regulatory-CMC nuance and the required expertise. It may present guidance without considering the clinical stage, product class, or regional nuances. If a prompt is developed by someone with no CMC expertise, it may lead to misleading, incomplete, or irrelevant outputs. Clear, context-specific questions-framed with an understanding of clinical stage, region, and technical nuance-are essential to guide the AI toward accurate and actionable responses. That’s where human expertise remains crucial.
In summary, AI is a valuable assistant-but not a substitute for CMC judgment. By combining AI’s efficiency with rigorous validation and oversight, we can use it responsibly and effectively in CMC consulting.
About the author
Tamar Oved
QA and CMC Director at ADRES Advanced Regulatory Services
Tamar has over fifteen years of experience in the pharmaceutical and biotechnology industry. She is experienced in quality assurance, quality control and manufacturing of drugs and biological products. She oversees GMP (Good Manufacturing Practices), GLP (Good Laboratory Practices) and CMC (Chemistry, Manufacturing and Controls) activities at production or testing sites. Tamar also has experience with aseptic processes. https://www.linkedin.com/in/tamar-oved-86082a1b/
From Insight to Access: How Integrated AI Solutions and Smarter Reimbursement Can Redefine Cancer Care
By Darron Segall MHS, is a market access leader
By Asher Nathan Venture Builder
Artificial intelligence is transforming oncology by enabling faster, more precise, and scalable approaches to cancer diagnosis and treatment. For decades, testing has largely relied on DNA mutation analysis—a process that is costly, time-intensive, and ultimately benefits only a small fraction of patients. In fact, fewer than one in five cancer patients have actionable DNA mutations, meaning the majority begin treatment without a targeted match, often relying on less tailored solutions.
Today, more advanced approaches are emerging. There are now emerging AI platforms that, by analyzing RNA expression by digital pathology images alone can identify treatment options for more than 90% of patients. Compared with DNA-only testing, which requires weeks to obtain a result and costs thousands of dollars, these newer methods deliver results in just days and at a fraction of the cost. The impact is significant: more patients receiving effective treatments, quicker therapeutic decisions, and fewer wasted efforts on ineffective therapies.
Yet diagnostics are only the beginning. The real transformation will occur when these AI technologies are integrated into top-tier end-to-end AI-based care continuum ecosystems. These systems do more than just generate a test result. They can help guide patients and providers through the entire journey: identifying evidence-based treatment options, alerting patients and clinicians when therapies fail, matching individuals to clinical trials, securing access to off-label or compassionate-use drugs, and navigating the complexities of insurance and prior authorization. Patients will be supported not only with clinical intelligence but also with practical tools such as reminders, financial aid guidance, symptom tracking, and live chat support—to ensure continuity of care and the best possible patient outcomes.
This integrated approach establishes a powerful feedback loop: every patient outcome feeds back into the system, enhancing its predictive precision and continuously improving treatment matching. As data accumulates across thousands of patient journeys, it evolves into a dynamic engine—driving stronger scientific insights, accelerating clinical trial recruitment, and enabling more efficient payer decision-making.
However, technology alone is not enough. If technology integration is one side of the coin, sustainable reimbursement is the other. Without the right reimbursement model(s), even the most effective solutions risk underutilization. Current frameworks often fragment or silo coverage into individual services—a test, a consult, a lab fee—without recognizing the full clinical and economic value of coordinated AI-enabled solutions. This approach risks undermining the adoption of technologies aimed at improving outcomes and reduce costs across the entire journey.
What is required is a solution-based reimbursement model that spans the full continuum of care. This means aligning payment with the total value delivered across the continuum, from diagnostic insights to treatment navigation, outcomes tracking, trial access, etc. Such models may include establishment of CPT codes for AI-enabled testing platforms, bundled payments that capture the full scope of services, and value-based pilots that tie reimbursement directly to outcomes such as survival, adherence, reduced hospitalizations. Early payer engagement (health plans, health systems, employers, etc.) and evidence generation will be critical components of a successful strategy.
AI has demonstrated its ability to revolutionize cancer testing by providing faster, more affordable, and more comprehensive results than ever before. Its integration into patient navigation, trial enrollment, and insurance support ensures that insights are put into action. The final step is ensuring reimbursement keeps pace with this new reality. By funding solutions rather than silos, healthcare systems can realize the full potential of AI in oncology: more patients receive the right therapy the first time, resources are used more efficiently, and outcomes are improved on a larger scale.
About the authors
Darron Segall
MHS, is a market access leader
Darron Segall, MHS, is a market access leader with 20+ years of experience shaping reimbursement, health economics, and payer strategies for innovative medical technologies. He has a strong track record of securing coverage and payment pathways that expand patient access while driving sustainable revenue growth. Known for visionary thinking and executional excellence, Darron helps companies navigate complex healthcare markets with clarity and impact. www.dreambighealth.com
Asher Nathan
Venture Builder
Asher Nathan, Venture Builder: Asher was a Managing Director of Paramount BioCapital, one of the most successful venture capital firms in the United States. He also served as Managing Partner of Zoticon Bioventures, and was CEO of a series of biotech companies, including NeoTx Therapeutics, a novel immunotherapy Company now in phase 2.
The AKT Awakening: Immunity Pharma’s Novel Approach to ALS and Neurodegenerative Disease
By Aviva Sapir Global PR and Comms Professional Specializing in Biotech and Health Tech
If there’s one thing I took away from speaking with Eran Ovadia, CEO of Immunity Pharma, it’s that success in biotech isn’t necessarily a field of eureka moments, it’s an accumulation of small wins, each one a hard-earned validation. “Every day, or every few days, I find a new proof point,” he told me. “Some new signal that we’re going in the right direction. That’s what keeps me going.”
And he’s been going for quite some time. A longtime entrepreneur with a background in health and science, Eran founded Immunity Pharma around a promising scientific discovery: a peptide that showed an unusual effect—it helped protect animals from radiation damage, even when administered post-exposure. “It wasn’t preventative. The primary damage had already happened,” he explained. “And it still worked, most of animals recovered. That’s when I knew we might have a true drug.”
The peptide was first developed by Professor Irun Cohen at the Weizmann Institute, who originally named it stressin,a nod to its effects on cellular stress. Eran quickly saw potential with much wider applications. He joined forces with Cohen and began exploring how the compound might be used to treat neurodegenerative diseases, which are often tied to cellular stress pathways.
After reviewing several disease candidates, the team chose to pursue the science in treating ALS, securing early preclinical funding from the Israeli ALS Association, IsrALS. Initial animal studies showed encouraging results, prompting the formal founding of Immunity Pharma. “We had a few options in parallel,” Eran said. “But the signal in ALS was strong, so we focused on it.”
IPL344 is now part of a family of peptides developed to activate the same stress-response mechanism. While ALS is the initial focus, the potential applications are extensive. “Today it is a platform,” Eran said. “We believe it could support a pipeline of therapies targeting other age-related diseases.”
Why ALS?
Amyotrophic lateral sclerosis (ALS) is a rare but devastating neurological disorder which causes progressive degeneration of motor neurons, leading to muscle weakness, difficulty speaking and swallowing, and eventually, respiratory failure. ALS is a highly challenging disease with a grim prognosis—the majority of ALS patients survive only two to five years after diagnosis, although life expectancy can vary greatly.
For a company like Immunity Pharma, ALS presented both a scientific challenge and an opportunity. “An advantage of focusing on ALS is that it’s one of the most aggressive diseases related to cellular stress,” Eran told me. “If it works there, we’ll know. And if it doesn’t, we’ll know that too. We won’t waste time and resources on futile dreams.”
That commitment to clarity shaped everything about Immunity’s lead candidate, IPL344. Most approved ALS therapies address symptoms or target a single process, such as oxidative stress or inflammation. IPL344 is designed to address six disease-driving mechanisms simultaneously, a distinction Eran believes is key.
The drug is a novel peptide designed to reactivate the AKT pathway, a crucial cellular signaling mechanism that supports neuron survival, reduces oxidative stress, and maintains metabolic stability. In ALS, this cellular pathway is often downregulated, and many drugs that attempt to stimulate it via cell membrane receptors fail because those receptors are themselves impaired.
Immunity Pharma’s approach is different. IPL344 bypasses the usual membrane receptor mediated pathways, instead entering the cell and activating a stress response that reawakens the AKT pathway from within. It’s a workaround, or as Eran put it, “a back door into the pro- survival system of the cell.”
That mechanism matters because ALS is a multi-pathology disease. “You’re not dealing with one problem,” Eran said. “You’re dealing with apoptosis, oxidative stress, misfolded proteins, disrupted metabolism, DNA damage, even immune dysfunction.” IPL344 addresses most of these, he believes, by unlocking AKT and, with it, the cell’s self-repair machinery.
Early results seem to back him up. In a Phase 1/2a open-label study, IPL344 was administered intravenously once daily, and preliminary data suggest about a 60% slower rate of disease progression in people living with ALS who received it, a rare, perhaps even unique, result. Additionally, the treatment was well tolerated and easily administered at home.
“We treated ten patients,” Eran said, “and we saw disease slow down in nine of them. Some were small, most of them significant, but there was a clear effect in multipledisease parameters.”
So, with these initial encouraging results, what are the next steps for the company?
Eran stressed that the company couldn’t have gotten to where it is today without the close partnership of scientists Prof. Irun Cohen, Dr. Ilana Cohen, and Dr. Oren Becker who cofounded the company with Eran, and the angels Rony Pfeifer, Gideon Stein and Yigal Tamir who joined him soon after, creating a small professional community around this endeavor. This core group has since expanded with dedicated clinical, scientific, professional, and business partners who have a shared vision and align with the business goals, maintaining long-term involvement in the company.
The Funding Journey
Like many early-stage biotech CEOs, Eran needs to spend a lot of his time and effort on fundraising. After good success with private investors, Immunity is now looking to include institutional partners to bring IPL344 to the finish line. That leap, from private to institutional backing, is particularly tough in Israel. “In the U.S., institutional investors are used to this stage. Here, it’s much harder to bring them in unless you are already past the finish line.”
If there’s a message here for other health tech founders, it might be this: storytelling matters. “Since I started the company, I’ve rewritten our presentation 200 times,” Eran said. While the science evolved gradually, the way he communicates it must constantly adapt to shifting business and scientific narratives. “You need to keep refining how you communicate the value of what you’re doing, maintaining a consistent core while demonstrating clear progress. That’s how you bridge the trust gap.”
Despite those hurdles, Eran stays focused. “This is a marathon, and we’re in the last ten kilometers,” he said. What helps him endure? A combination of incremental wins and belief in the mission of helping stop the progress of dreaded neurodegenerative diseases.
As Immunity Pharma prepares for the next stage, Eran is clear-eyed about what lies ahead. “In five years, I hope we will have a drug on the market. I hope we’ve expanded the platform to treat other neurodegenerative diseases. But most of all, I hope we’ve helped people.”
It’s that final point—the patients—that keeps coming back in our conversation. “I know this drug helped the people who took it. I saw it,” he said. “Now we need to prove it, at scale. That’s the mission.”
Immunity Pharma began collaborating with ADRES.bio in 2012, advancing the company’s groundbreaking work to develop a treatment for ALS. Early in the partnership, ADRES.bio supported the production of the first manufacturing batches, enabling compassionate use treatment for a patient whose life was extended by 18 months. Over the years, the two teams have worked closely to navigate the complex regulatory path toward approval, sharing in the progress and discoveries that continue to reveal the therapeutic potential of Immunity Pharma’s product.
About the author
Aviva Sapir
Global PR and Comms Professional Specializing in Biotech and Health Tech
Aviva Sapir is a global PR and comms professional specializing in biotech and health tech, currently serving as Head of HealthTech Communications at Number 10 Strategies. Aviva leverages deep media relations expertise to help companies build credibility and secure recognition in the market. Number 10 Strategies offers a full range of PR and communications services across multiple industries. https://www.linkedin.com/in/aviva-sapir-pr-and-marketing/
Beyond Vessel Occlusion. A Porous-Microsphere Approach to Reinventing TACE in HCC
By Aviva Sapir Global PR and Comms Professional Specializing in Biotech and Health Tech
Liver cancers are one of the most common cancers worldwide and a leading cause of cancer mortality, and numbers are only rising globally. The standard treatment for intermediate-stage hepatocellular carcinoma, transarterial chemoembolization (TACE), treats approximately 40% of liver cancer patients, yet the outcomes remain grim: nearly half of those who receive TACE die within two years of diagnosis, and only about one in five survive beyond five years. This sobering prognosis highlights why TACE represents one of oncology’s most critical unmet needs.
That’s where BDUK Therapeutics, an Israeli biotech startup poised to redefine TACE, comes into the picture. Co-founders Walter Wasser, MD and Yaron Suissa, PhD, MBA, contend that the fundamental flaw isn’t in the concept of targeting tumors by disrupting their blood supply, but rather in current methods that completely occlude vessels, inadvertently preventing the very chemotherapy they aim to deliver from reaching its cancerous target.
Their answer is EmboPore, a novel porous microsphere-based delivery technology developed by Professor Ofra Benny at Hebrew University, which is designed to maintain partial blood flow, allowing chemotherapy to be delivered directly to tumors over a sustained two-week period. Recent advancements in the field, showing improved progression-free survival when TACE is combined with targeted and immunotherapy drugs, further highlight the urgent demand for more effective TACE platforms—a need BDUK Therapeutics is uniquely positioned to address.
With approximately 900,000 new hepatocellular carcinoma diagnoses globally each year, and the TACE market alone valued at $11.8 billion in 2024 and projected to reach $16.4 billion by 2031, the moment for this innovation could not be more opportune. We recently sat down with BDUK co-founders Walter and Yaron to learn more about their approach to this immense challenge.
How did the idea for BDUK Therapeutics come together, and what made this particular approach to liver cancer stand out to you?
Walter: Yaron and I had been exploring multiple biotech opportunities, looking for something that addressed a major clinical gap. As a physician and a scientist, we were drawn to projects that could make a real difference.
After reviewing around ten technologies, we found Professor Benny’s work on porous microspheres designed to treat liver cancer especially compelling. The science was strong, the need was urgent, and it was clear this product had the potential to change the standard of care. That’s when we decided to build BDUK Therapeutics around it.
Liver cancer accounts for more than 700,000 deaths annually worldwide, and TACE remains the standard of care for many patients. Why does TACE continue to fall short, and how does that shape the opportunity for innovation?
Walter: Liver cancer is a major global health challenge. The Lancet Commission’s July 2025 report on global hepatocellular carcinoma has already garnered significant attention, identifying liver cancer as the sixth most common cancer worldwide and third leading cause for cancer-related mortality globally. The commission projects new liver cancer cases will nearly double from 870,000 in 2022 to 1.52 million by 2050.
There are 60,000 to 70,000 new cases in the U.S. and Europe each year, with only 40% of these individuals are eligible for curative surgery. Another 20% have advanced disease with no treatment options. This leaves 40% of patients to be treated with TACE, transarterial chemoembolization.
TACE involves injecting beads mixed with chemotherapy into the hepatic artery to block blood flow and target the tumor. In theory, this starves the cancer and delivers the drug directly. But in practice, the occlusion is so complete that very little of the chemotherapy reaches the tumor. That’s the fundamental flaw.
As a result, outcomes remain deeply concerning. Nearly half of the patients who receive TACE die within two years of diagnosis, and only about one in five survive beyond five years. These are not just statistics; they reflect the limitations of current therapies and the urgent need for better options.
Your solution, EmboPore, was designed to overcome the core limitations of conventional TACE, namely, poor drug delivery and irreversible vessel occlusion. Can you explain how it works, and why it represents such a significant advance over the current standard of care?
Walter:EmboPore fundamentally reimagines how locoregional therapy is delivered. Traditional TACE uses embolic beads to block the hepatic artery and starve the tumor, but the complete occlusion means that very little of the chemotherapy actually reaches the cancer. That’s the root problem.
What Professor Benny developed is a porous microsphere that allows partial blood flow to continue, enabling embedded drugs —Doxorubicin and Tirapazamine (TPZ)—to reach the tumor over an extended two-week period. These microspheres are made of PLGA, a biodegradable polymer that rapidly breaks down in acidic environments, which are the conditions typically found in tumors. After an initial rapid degradation, the microspheres degrade in a gradual manner. This is a controlled degradation that releases the two drugs gradually and directly into the tumor microenvironment.
There is also a synergy between the two drugs: doxorubicin is a standard chemotherapeutic agent, while TPZ is only activated by hypoxia. The microspheres themselves induce localized hypoxia, activating TPZ which together with DOX provide the synergistic effect. Because EmboPore eventually dissolves, repeat treatments through the same arterial portal become a possibility—something conventional TACE doesn’t allow. The inability to re-catheterize the same hepatic artery has a significant negative impact on liver burden and on patients’ ability to undergo repeat procedures.
You can think of it like a drug-eluting stent, but for liver tumors: precise, sustained drug delivery at the site of disease with minimal systemic toxicity while enabling the required environment for optimal anti-tumor drug impact. In preclinical models, this resulted in Significantly higher drug concentrations within tumors, with minimal drug exposure in the surrounding liver tissue.
From a pharma R&D and business development perspective, what makes this platform fundamentally different from other attempts to improve TACE?
Yaron: The key limitation in TACE is the complete blockage of blood vessels, which not only prevents drug penetration but also limits the potential for repeat dosing. The field has optimized everything about the beads—size, material, drug binding—except the main problem: they completely occlude the vessel.
Our product solves that. The porous structure of the microsphere allows some blood to continue flowing through, improving drug exposure to the tumor and preserving vessel patency. It degrades faster in acidic environments like the tumor microenvironment, which further enhances drug release at the right time and place. It’s a delivery solution that works with the biology, not against it.
The TACE market is estimated at billions of dollars—how do you evaluate commercial potential and how does orphan drug designation factor into your strategy?
Yaron: The commercial potential is substantial. In 2023, the TACE market reached $11 billion globally, with over 1.3 million patients eligible for treatment. While the largest patient populations are in Asia, there are still 60,000 to 70,000 eligible patients each in the U.S. and Europe.
Given the patient population size for this condition, we anticipate qualifying for orphan drug designation in both the U.S. and Europe, which would unlock significant incentives such as market exclusivity, fee reductions, and development support.
We also believe we may be eligible to receive FDA breakthrough therapy designation (BTD), and PRIME status in Europe, which can accelerate regulatory review and potentially enable commercializing the drug before completing Phase 3 trials. From a business perspective, the market average for orphan drug ROI is compelling—even under conservative models the return is projected to reach 7x.
Additionally, we are currently observing a significant change in market trends. Whereas in the past startup interactions with big pharma were initiated only towards the end, or after completion of, a phase-2 clinical trial, it’s been reported that leading pharma companies are now investigating M&A deals much earlier, even after phase-1 studies with positive readout.
What’s your current funding timeline and how are you structuring the path to clinical trials?
Yaron: We’re in the process of raising funds to start Phase 1 over the next 18 months. That includes completing GLP-compliant large animal studies and initiating first-in-human trials with secondary efficacy endpoints. We’ve already started some preparatory work.
Interestingly, two of our three components, PLGA and Doxorubicin, are FDA approved, while the third, TPZ, is well known and previously tested. The vast existing safety data may allow us to expedite traditional preclinical steps and skip non-rodent toxicity studies or run them in parallel with the clinical study to save time.
While your lead indication is liver cancer, do you see broader applications for this platform beyond hepatocellular carcinoma (HCC)?
Yaron: Absolutely. While our initial focus is on primary liver cancer, EmboPore is equally relevant for secondary liver tumors—cancers that metastasize to the liver. These include colorectal, pancreatic, breast, and uveal melanoma, which often spread preferentially to the liver and are difficult to treat systemically.
The strength of EmboPore lies in its ability to deliver various chemotherapy locally, directly into the tumor’s blood supply, while preserving vessel patency. This allows for sustained drug exposure, reduced systemic toxicity, and the possibility of repeat treatment, thereby overcoming major limitations of conventional TACE.
Looking ahead, we also see potential applications in other solid tumors beyond the liver. Any cancer accessible through the arterial system—like kidney, prostate, or even brain tumors such as glioblastoma—could benefit from a targeted, locoregional drug delivery approach. While we’re not currently targeting non-oncology indications, the underlying technology may also have future relevance in areas like arterial inflammation or vascular diseases.
You’ve received some strong early interest from key clinical opinion leaders. How is that helping to shape your path forward?
Yaron: It’s been a strong indicator that we’re solving the right problem. Usually, it takes months to get KOLs to engage meaningfully, but we’ve had experts in the field want to get involved almost immediately.
We had a leading senior hepatologist interested in collaboration after presenting our supporting scientific data. We’ve also had interest from a translational oncology expert at a leading cancer center in New York as well as leading senior interventional radiologists both in Israel and the US. That kind of engagement, from individuals with deep experience in liver-directed therapies, confirms that we’re addressing a significant clinical gap.
We are confident that our mission to transform the treatment paradigm for individuals with intermediate-stage liver cancer will have far-reaching implications for patients worldwide, offering new hope and meaningful advances in care.
For BDUK Therapeutics Ltd., the path ahead means proving their porous microsphere technology can succeed where standard TACE has failed, and clinical trials will provide the answer. With high liver cancer mortality rates and a clear market waiting, BDUK Therapeutics offers both hope for patients and potentially a blueprint for how academic research can be translated into life-saving therapies.
ADRES Bio is actively supporting BDUK Therapeuticson their regulatory path, including with CDMO selection, CMC activities, preclinical plans, grants, investor meetings, and regulatory discussions.
About the author
Aviva Sapir
Global PR and Comms Professional Specializing in Biotech and Health Tech
Aviva Sapir is a global PR and comms professional specializing in biotech and health tech, currently serving as Head of HealthTech Communications at Number 10 Strategies. Aviva leverages deep media relations expertise to help companies build credibility and secure recognition in the market. Number 10 Strategies offers a full range of PR and communications services across multiple industries. https://www.linkedin.com/in/aviva-sapir-pr-and-marketing/
By Roy Zaibel Co-CEO at ADRES and Chief Editor of the Bio-Startup Standard
Let me start with the obvious. AI has dominated the conversation for the last three years, and as the editor that creates a real dilemma. When everything is labeled AI, an AI issue can feel predictable before you turn the first page. I did not want that. I wanted this issue to be useful and grounded, to give you practical takeaways you can apply with your team, and to spotlight a few unusual applications that challenge how we think about the work.
On the clinical side, we move past the usual headlines and get into design choices. One feature examines digital twins that can right-size control arms without compromising statistical power, which is a practical lever on timelines and budgets. Digital Twins in Clinical Trials. Another looks at the EU AI Act and what it means when your trial relies on systems that regulators consider high risk; the takeaway is straightforward. If AI touches regulated work, build for transparency, documentation, and human oversight you can defend in an audit. EU AI Act and Clinical Trials. To balance optimism with discipline, we include a perspective on where AI helps regulatory planning today and where it does not, so you can set credible expectations with your stakeholders The Future of AI in Regulatory Planning: Progress with Caution.
To help you operationalize all this, we include a compact toolkit for AI governance and traceability so you can measure, explain, and reproduce model behavior in environments that regulators actually understand ==[link: Toolkit: TruLens, Weights & Biases, ClearML]==. And we step outside software with a piece on protein design that starts from nature’s own blueprint and moves toward formulations that hold up in the real world; different field, same principle of starting from what works and building carefully ==[link: Human Milk as Nature’s Gold Standard]==.
My goal with this issue is simple. Less noise and more signal. If you are building, testing, or scaling in biotech or medtech, I hope these stories help you move faster and safer. Our role at ADRES.bio is to make that practical; we help teams turn AI into inspection-ready operations with people who know the science and the systems. If something here sparks a question, tell us. And if you have a case study for the next issue, send it our way.
About the author
Roy Zaibel
Co-CEO at ADRES and Chief Editor of the Bio-Startup Standard
Roy helps non-EU startup companies obtain their SME status
From Code to Clinic: Navigating the Human-Machine-Doctor Triangle in AI-Driven Medicine
By Elad Levy Lapides CEO, DrugsIntel | CEO, Global Precision Medicine & Tech, SK Pharma | CEO, SKcure
Exploring Challenges and Approaches for Ethical and Effective AI in Healthcare
The rapid growth of health data from electronic health records, genomics, and wearable devices has created unprecedented opportunities in medicine. Artificial Intelligence is increasingly viewed not merely as a support tool but as a cornerstone for future diagnostics and personalized treatments. This shift, while promising, introduces significant challenges: ensuring transparency, preventing bias, and integrating technology with human clinical judgment.
One promising approach gaining traction is phenotype-driven medicine. Instead of grouping patients by broad disease labels, this method identifies smaller, more meaningful clusters based on biological markers, behaviors, and health histories. Such clustering can guide highly personalized treatments and improve outcomes compared to traditional, one-size-fits-all models.
A practical illustration of this can be found in initiatives like Obesio, which applied phenotype clustering to weight management. By identifying six distinct metabolic profiles, Obesio enabled tailored interventions that combined medication, nutrition, and exercise specific to each profile. Results from early pilots showed over 30% greater weight loss and improved patient adherence compared to conventional methods, highlighting the potential impact of this approach.
Introducing AI tools into clinical workflows requires more than technological sophistication; it demands careful consideration of ethics and operational realities.
A useful framework to guide this integration is the Human-Machine-Doctor Triangle, which focuses on three critical dimensions:
Explainability (XAI): Physicians must understand why an AI makes a recommendation. Transparent models help build trust and support informed clinical decisions.
Bias and Equity: AI models must be trained and audited with diverse data to avoid reinforcing existing health disparities. Ongoing evaluation across populations is essential.
Data Privacy and Patient Autonomy: Trust is earned through transparency and compliance with global standards like GDPR and HIPAA, ensuring patients know how their data is used and protected.
These principles aim to ensure AI enhances rather than replaces the human elements of care-empathy, judgment, and the patient relationship. When implemented thoughtfully, AI can surface insights from data too complex for humans to process, allowing clinicians to focus on what they do best: caring for patients.
As healthcare moves toward proactive, predictive, and participatory models, frameworks like phenotype-driven medicine and the Human-Machine-Doctor Triangle offer practical pathways forward. They demonstrate that the future of personalized medicine will rely not just on technology, but on the careful blending of computational power, ethical oversight, and clinical wisdom.
About the author
Elad Levy Lapides
CEO, DrugsIntel | CEO, Global Precision Medicine & Tech, SK Pharma | CEO, SKcure
Elad Levy Lapides is a global executive specializing in precision medicine and AI-driven healthcare. He has extensive experience in leading pharmaceutical innovation, supply chain excellence, and strategic partnerships. His focus is on transforming healthcare through innovation, efficiency, and collaboration.
We use cookies to enhance your browsing experience and analyze our traffic. By continuing to use our site, you consent to our use of cookies.
Learn more